



Why alternative splicing?
One enduring question is how a genotype contributes to a phenotype. We have seen dramatic advances in high-throughput technology, and high-throughput studies of biological systems are rapidly accumulating a wealth of 'omics'-scale data. The development of Next Generation Sequencing technology is rapidly changing the face of the genome annotation and analysis field. We are now able to use genome sequence and mRNA expression data to improve our understanding of the pathogenic phenotype of human diseases or complex traits. There is a biological mechanism to relate the genome to the transcriptome. Short-term goal is to characterize this biological mechanism between these data that connect genotype to phenotype by focusing on alternative splicing (AS). Long-term goal is to create a molecular picture for genomics and personalized medicine. Our previous works have already developed the computational pipeline for integrating genomics with transcriptomics and providing functional annotation for intragenic SNPs involved in splicing regulation. The prominent works include incorporating these element into i) study of genetic basis of variations that affect splicing in human populations, ii) crosstalk between epigenetics and AS for exon recognition, and iii) resource generation for scientific communities. These resources harness the power of genome variation that facilitates enhanced understanding of its contribution to health disparities for diseases.
Multi-omics data integration tools
Genome-wide research has generated various data including multiple genome, transcriptome, epigenome, microRNAome, and proteome data, making it possible to conduct an integrative omics analysis. There exists clear recognition that the utilization of these multi-layered omics data is highly informative in understanding the complexity of RNA regulation. Therefore, we develops general resources that provide mechanistic information between DNA sequences and phenotypes through RNA regulation.
IMAS Integrative analysis of multi-omics data for alternative splicing
https://bioconductor.org/packages/release/bioc/html/IMAS.html
CAS Visualization of Cancer Alternative Splicing
http://genomics.chpc.utah.edu/cas/
ADAS Visualization of Alzheimer's disease Alternative Splicing
Lims
In the wake of the coronavirus disease 2019 (COVID-19) pandemic, there has been a significant increase in attention and research focus on infectious diseases. This has led to the emergence of novel infectious diseases as a potential future threat. While Laboratory Information Management Systems (LIMS) were initially developed to facilitate rapid non-clinical drug testing for COVID-19, the post-pandemic research landscape has necessitated a shift towards understanding the underlying biological mechanisms of the infectious disease. Therefore, it is a nature growing a need for LIMS to encompass research on other infectious diseases and to accommodate various research protocols.
To address these challenges, we propose advanced version of LIMS (LIMS version 2) that accommodates a flexible and expandable LIMS capable of managing diverse research protocols and standardizing the organization of vast amounts of data. The advanced system incorporates enhanced user authority levels and implements a robust security system to ensure data privacy through encryption measures. A relational database management system (RDBMS) was also developed to efficiently store and manage the structured data.
A report tool was further developed to support multiple file formats (MS Office, HWP, and PDF), streamlining the reporting process and facilitating effective communication of research findings. Moreover, the new component, facilitates the management of stock for model organisms, offering convenient accessibility to the director responsible for overseeing this aspect.
Our LIMS version 2 represents a significant advancement in infectious disease research by providing researchers with a comprehensive platform that supports the investigation of multiple infectious diseases and diverse research protocols. The system's flexibility and expandability enable it to adapt to evolving research needs, making it an invaluable tool for studying disease mechanisms. By promoting features optimized for non-clinical research, our LIMS version 2 promote researcher not only to continue scientific study and collaboration in post-pandemic era but also to quickly respond to the future pandemic threat.

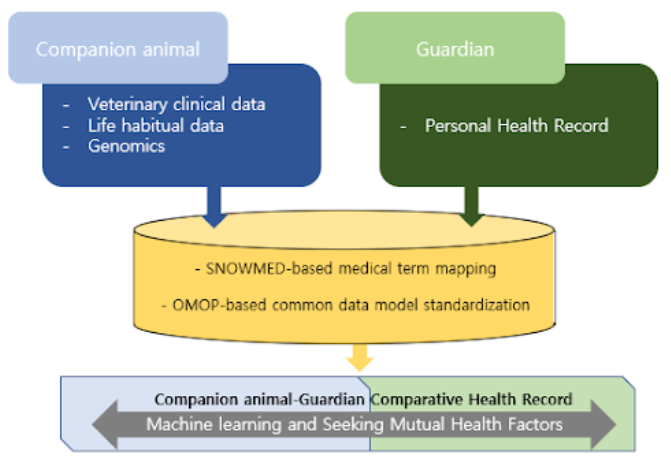
Human-Animal
This project aims for the production, collection, and analysis of preclinical data from infectious disease studies to establish and utilize the national infrastructure for responding to infectious diseases including COVID-19 and future Disease X. Laboratory Information Management System (LIMS) is a software-based solution for the operation of laboratories. Our project is developing a LIMS optimized for national preclinical testing. We're developing an open portal (website) to display the findings from preclinical testing. The data collected in LIMS will be visualized and shown on the portal to offer a potential resource for researchers in the field of infectious disease studies.

Background
Supervised machine learning models have been widely utilized for prediction and getting insight into diseases by classification of patients based on personal health records. However, a class imbalance is an obstacle that disrupts the training of the models.
Objective
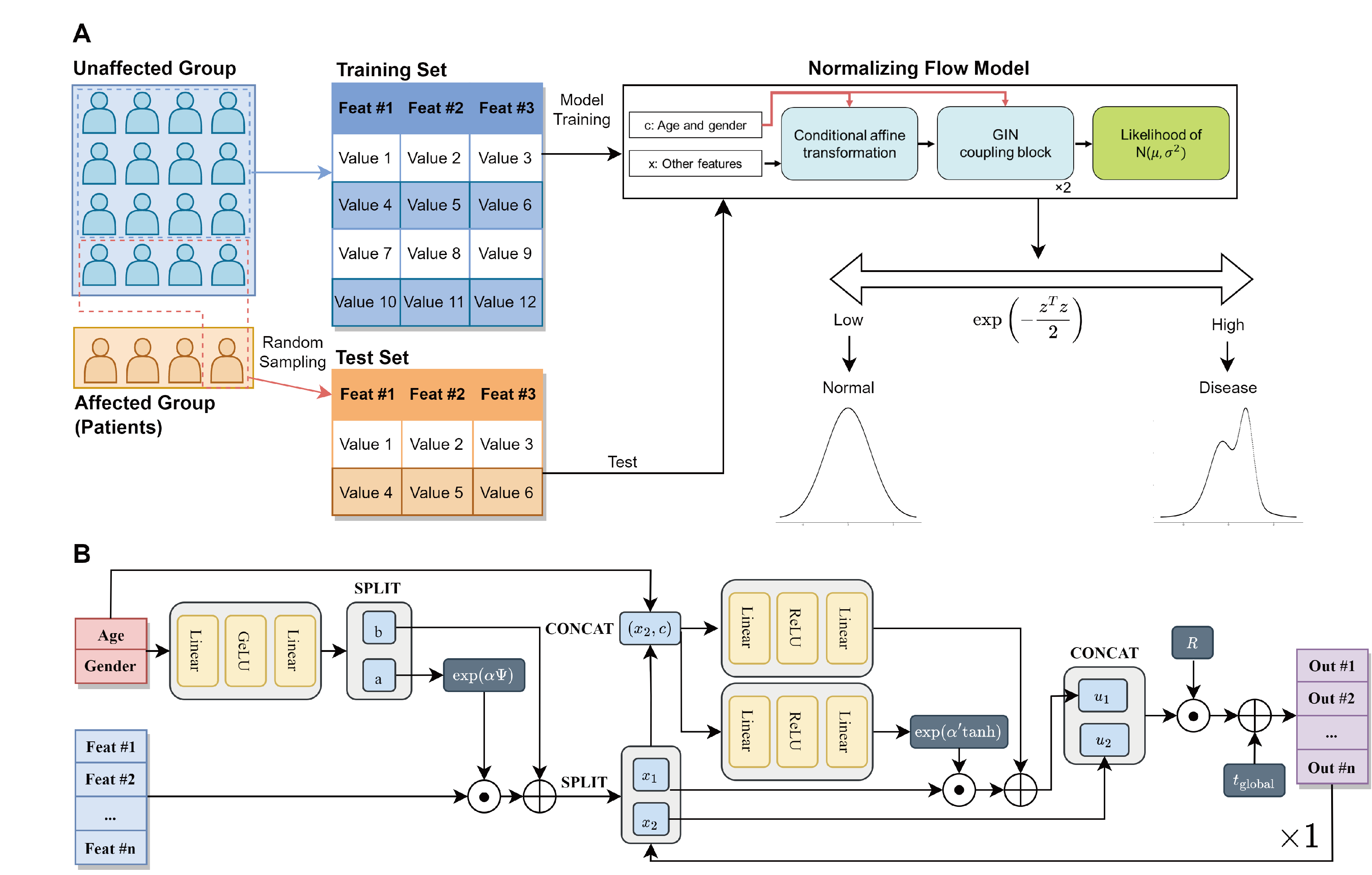
We aimed to address class imbalance with a normalizing flow, one of the semi-supervised models for anomaly detection. It is the first introduction of the algorithm for tabular clinical data.
Methods
We collected data from 706 South Korean citizens, featured with genetic data obtained from direct-to-customer service (microarray chip), medical health check-ups, and lifestyle log data. Based on the health check-up data, six chronic diseases were labeled (obesity, diabetes, hypertriglyceridemia, dyslipidemia, liver dysfunction, and hypertension). After preprocessing, supervised classification models and semi-supervised anomaly detection models, including normalizing flow, were evaluated for the classification of diabetes, which had extreme target imbalance (about 3%), based on AUROC and AUPRC. In addition, we evaluated their performance under the assumption of insufficient collection for patients with other chronic diseases by undersampling disease-affected samples.
Results
While LightGBM (the best-performing model among supervised classification models) showed AUPRC 0.155 and AUROC 0.832, normalizing flow achieved AUPRC 0.326 and AUROC 0.819 during ten evaluations of the classification of diabetes. Moreover, normalizing flow could outperform the supervised model under a few disease-affected data numbers for the other five chronic diseases.
Conclusions
Our research suggests utilization of normalizing flow when cases are insufficient for prediction of chronic diseases with personal health records.
SplicingML
Genome-wide association studies (GWAS) have discovered enormous genetic variants associated with complex disease, including Alzheimer’s disease (AD), and these variants have contributed to a development of candidate preventive strategy and early detection biomarkers. Integrative analysis of multi-omics data plays a significant role in advancing our understanding in functional mechanisms of the AD-associated SNPs from GWAS. Through integrative analysis of genomic and transcriptomic data, disease-associated SNPs has been considerably reported as splicing quantitative trait loci (sQTLs) that affect alternative splicing exon alterations. However, most of GWAS studies have generated and analyzed genomic data, having not co-ascertained RNA-seq data, and it limits us to fully explore molecular mechanisms of GWAS-based disease-associated SNPs. In this study, we aimed to develop the predictive model that identifies genetic variants (sQTLs) affecting AS pattern changes from only genomic data by using machine learning techniques that have successfully allowed us to construct high accurate predictive models.

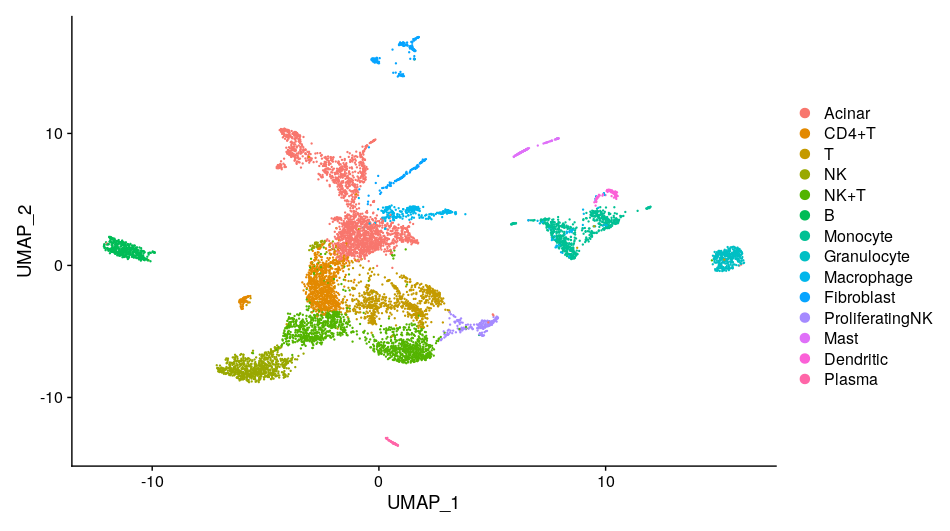
scRNA-seq
Single-cell RNA sequencing (scRNA-seq) technology is a revolutionary technique that allows scientists to understand the transcriptome at the individual cell level. This advanced method has emerged as an indispensable tool in genomics, enabling researchers to unravel the complexities of cellular diversity and function.
One of the central strengths of scRNA-seq lies in its ability to unveil the inherent heterogeneity of cellular populations. Traditional bulk RNA sequencing measures the average expression of genes across thousands to millions of cells. While it provides valuable insights, it often overlooks the contribution of individual cells, masking unique or rare cell types and states. scRNA-seq, in contrast, allows us to peek into this unseen diversity, illuminating a comprehensive landscape of cellular identities and states.
The application of scRNA-seq is wide-ranging, stretching from basic biology to medicine. For instance, in developmental biology, it can trace the trajectory of cell differentiation and the genesis of various cell types from a single progenitor. In neuroscience, it is used to delineate the vast heterogeneity of neurons and glial cells. Meanwhile, in oncology, it can uncover the tumor microenvironment, revealing not only the tumor cells' subclonal structure but also the spectrum of immune and stromal cells involved in tumor progression and response to therapies.
Our project aims to Analysis characteristics and inhibition mechanism of tumor microenvironment (TME) through the study of interaction between pancreatic cancer and cancer associated fibroblast (CAF).

standard cohort
The aging population in South Korea is rapidly increasing. In 2015, the elderly population accounted for 13.1% of the total population, and it is projected to reach around 40% by 2060. This global trend of aging is also leading to a rapid increase in the number of dementia patients. According to a survey on dementia prevalence conducted in 2016, there were 750,000 dementia patients among the population aged 65 and older in 2018. This number is estimated to reach 1 million by 2024 and exceed 2 million by 2039. Consequently, there is a pressing need for research on the causes and treatment methods of dementia.
In the study titled "Anticholinergic Drug Exposure and the Risk of Dementia: A Nested Case-Control Study," the researchers investigated the association between the use of anticholinergic drugs and the increased risk of dementia in patients diagnosed with dementia compared to a control group without dementia. Specifically, the study focused on a sample of 58,769 dementia patients and 225,574 individuals in the control group registered in the QResearch primary care database in the United Kingdom. Furthermore, there are ongoing research efforts in various international studies to explore the link between anticholinergic drugs and dementia.
Therefore, we intend to conduct a study using Korean cohort data to examine the association between anticholinergic drugs and dementia. The results of this research will be published on our official website.
AiChatVet
This project aims to provide information by predicting the health status and suspected diseases of visiting patients based on EMR data from veterinary hospital. AI-ChatVet is an app that provides an enhanced consultation experience for owners and provides urgency level by predicting diseases. This project algorithmically implements the correlation between diseases and EMR data. The results of the consultation and the predictions from the algorithm can be used to assist veterinarians in their practice.
